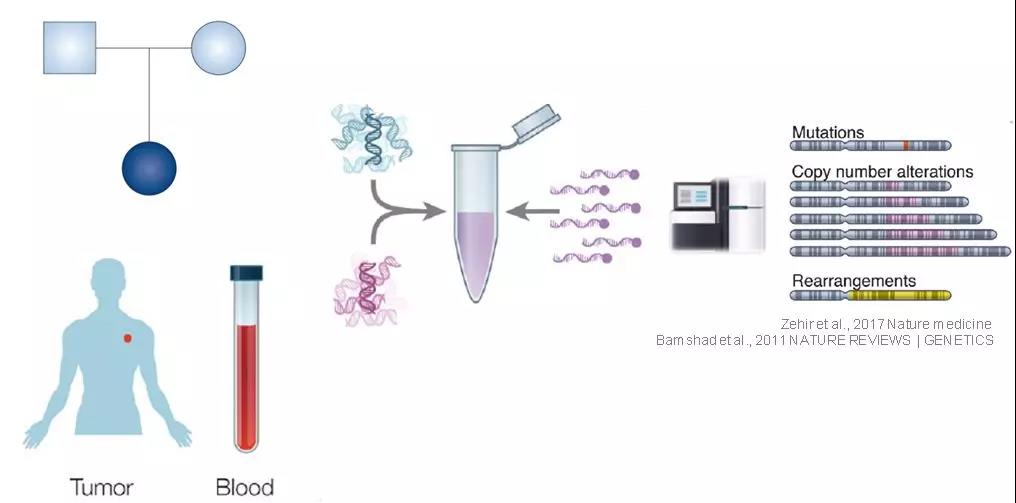
二代測序(NGS)的快速發(fā)展加速了遺傳病,、癌癥等領(lǐng)域的研究,其本身也作為一種更為先進的基因檢測手段逐步走進臨床,。在NGS的眾多技術(shù)中,,靶向測序正在成為新時代的“寵兒”。遺傳病診斷(WES/Panel),、腫瘤伴隨診斷(腫瘤多基因Panel),、免疫治療療效預(yù)測(WES/Panel),這些NGS應(yīng)用熱點都是靶向測序打下的“江山”,。今天,,咱們不談常規(guī)操作,讓小編給大家分享一些靶向測序的其他用途,。
研究人員發(fā)現(xiàn),,從HPV感染到癌變的過程中,有些HPV病毒基因會“嵌入”(整合)到人體細胞的基因組中,,整合事件的發(fā)生和癌變結(jié)局有著一定的相關(guān)性,,可以進一步作為分子標志區(qū)分高危人群。如何檢測這些入侵人類基因組的HPV呢,?2015年,,我國學(xué)者創(chuàng)造性地借助靶向測序來探究人類基因組上HPV的整合熱點和可能的整合機制。該研究發(fā)表在《自然-遺傳學(xué)》上,,堪稱靶向測序應(yīng)用的經(jīng)典之作 [1],。
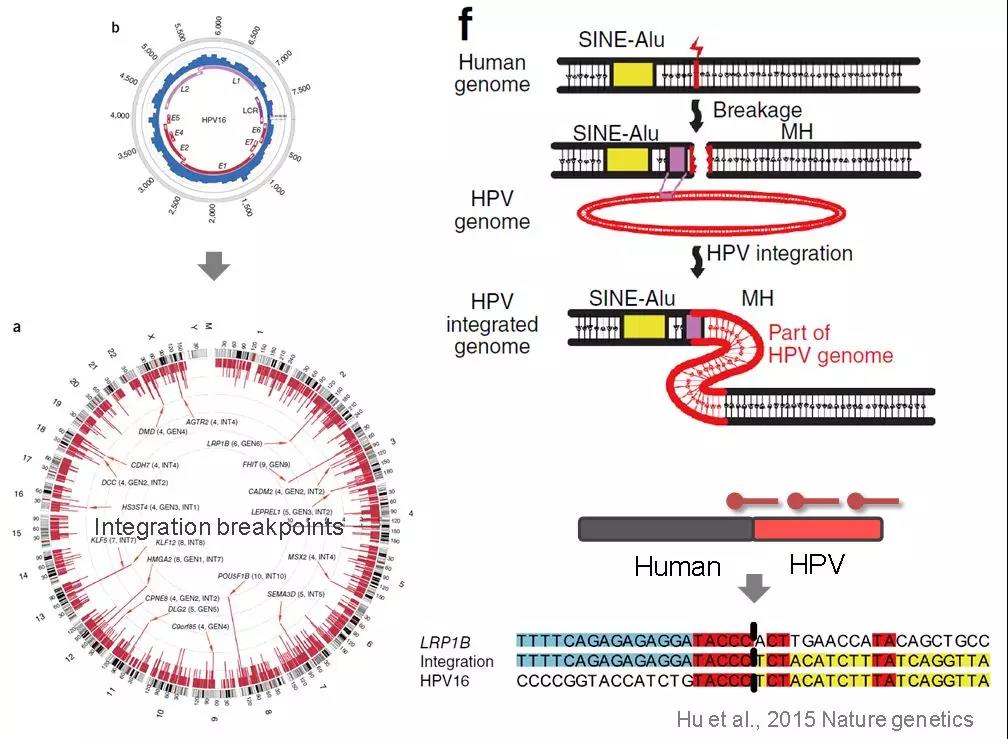
如圖2所示,根據(jù)HPV基因組序列設(shè)計探針,,從感染細胞中捕獲HPV基因組,,如果有整合發(fā)生,測序Reads中將同時存在HPV和人類基因組的序列,,與參考基因組比對,,便可獲得整合位置的信息。在利用細胞系樣品進行的性能驗證中,,靶向測序較WGS更加靈敏,,不但可以鑒定到WGS發(fā)現(xiàn)的絕大部分整合位點(10/11),,還額外檢測到135個WGS沒有檢出的位點。HPV的整合熱點可以作為宮頸癌患者早期個性化治療和進展評估的重要分子標志物,。
病原體基因組研究和臨床檢測
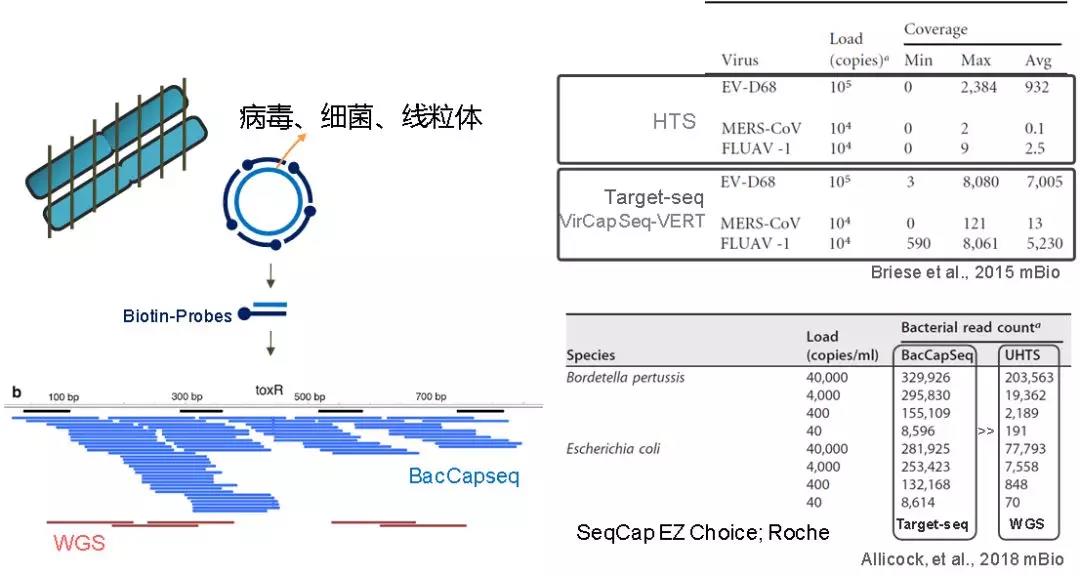
伯氏疏螺旋體(Borrelia burgdorferi)以蜱為主要傳播媒介,感染人體后會引起以神經(jīng)系統(tǒng)損傷為主的萊姆病,,據(jù)說紅極一時的搖滾歌手艾薇兒就因患此病退出歌壇多年,。從宿主中快速而準確地檢測人類病原體的全基因組序列,將有助于對病原體的流行病學(xué)和進化動力學(xué)進行精細解析研究,。但是,,在提取DNA之前較難事先分離宿主細胞和病原體,宿主基因組將造成大量的數(shù)據(jù)浪費,。以伯氏疏螺旋體為例,,它的基因組大小約為0.0015 Gb,而其宿主蜱蟲的基因組長約2.1Gb,,如果采用WGS進行研究,,伯氏疏螺旋體的有效數(shù)據(jù)可能只有可憐的千分之一(基因拷貝:1:1)。2015年,,有研究利用基于雜交捕獲的靶向測序技術(shù)富集蜱蟲體內(nèi)的伯氏疏螺旋體基因組,,數(shù)據(jù)有效率在70%以上(Roche SeqCap EZ Choice,[2]),。
傳染病的病原體鑒定,,細菌耐藥譜分析是NGS在臨床中的重要應(yīng)用方向。2013年《新英格蘭醫(yī)學(xué)雜志》發(fā)表的一篇報道中,,應(yīng)用全基因組測序,,可以將結(jié)核分枝桿菌耐藥譜檢測時間從幾個星期縮短到幾天,但昂貴的成本和檢測靈敏度限制了該方法的廣泛應(yīng)用[3],。面對這些挑戰(zhàn),,我們來看看捕獲測序的表現(xiàn)。VirCapSeq-VERT技術(shù)是針對病毒基因的NGS靶向檢測方法,,它包含約200萬條長度為100nt的探針(Roche SeqCap EZ Choice),,可在血清、血液和組織等復(fù)雜的樣本背景下,,檢測207種已知脊椎動物病毒[4],。相對于傳統(tǒng)的NGS技術(shù),VirCapSeq-VERT得到的Reads深度可提高100~10000倍,。此外,,還有前段時間報道的BacCapSeq技術(shù),可靶向檢測細菌基因組,。該系統(tǒng)包括420萬條長約75nt的探針(Roche SeqCap EZ Choice),,能夠一次性檢測307種細菌,,包括所有已知致病菌,抗生素耐藥基因和毒力因子,,其準確性與針對性細菌篩查相當(dāng),,而且檢測范圍更廣。要知道,,多重PCR一次最多檢測19種致病菌,。此外,BacCapSeq技術(shù)還更加靈敏(富集1000倍),,并且可以在70小時內(nèi)完成抗生素耐藥性檢測[5],。除應(yīng)用于病毒和細菌外,靶向測序技術(shù)也適用于線粒體DNA的突變檢測,。
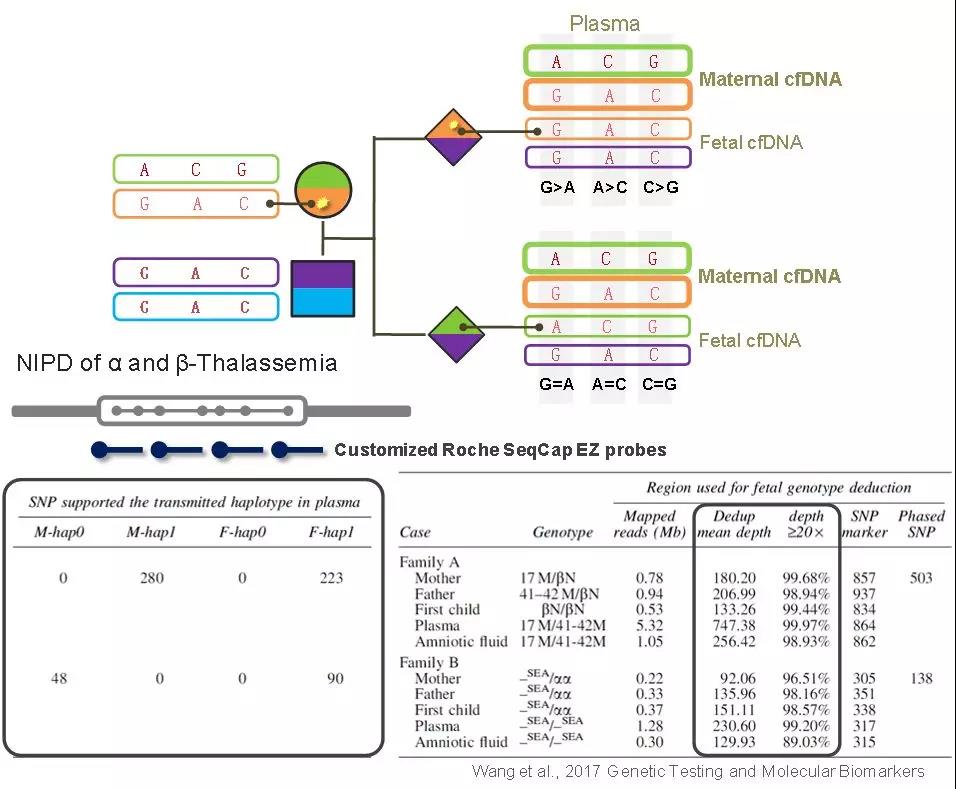
NIPD的檢測難點主要是在母親作為攜帶者的情況,,在孕婦的游離DNA中,母體基因型片段占據(jù)絕大部分,,只通過變異位點本身的頻率很難準確評估胎兒的基因型,。RHDO通過成百上千個“等位基因關(guān)聯(lián)的SNP”來提高準確性,并將胎兒基因型預(yù)測簡化為判斷胎兒遺傳了父母雙方的某種單倍型,。此外,,由于RHDO并不是直接檢測突變本身,所以,,無論致病變異是點突變還是大片段缺失,,RHDO均可勝任。如圖4所示,,在α和β地貧的無創(chuàng)產(chǎn)前診斷研究中,,先獲得父母雙方的單倍型信息,并對等位基因關(guān)聯(lián)的單倍型區(qū)域設(shè)計探針(Roche SeqCap EZ Choice),,然后對相應(yīng)區(qū)域的cfDNA進行捕獲測序,,富集單倍型區(qū)域,統(tǒng)計關(guān)聯(lián)SNP的頻率,評估整個單倍型區(qū)域的相對劑量,,最后以此預(yù)測胎兒基因型,。
基于芯片的GWAS可以發(fā)現(xiàn)復(fù)雜疾病顯著關(guān)聯(lián)的變異位點,,但多在非編碼區(qū),很難真正確定致病根源,?;谕怙@子測序的GWAS則可以將關(guān)聯(lián)變異位點鎖定在編碼區(qū)內(nèi),能夠為致病機制的闡明,,疾病的治療提供更豐富的線索,。
2. 新的身份:多基因高通量檢測的“金標準”
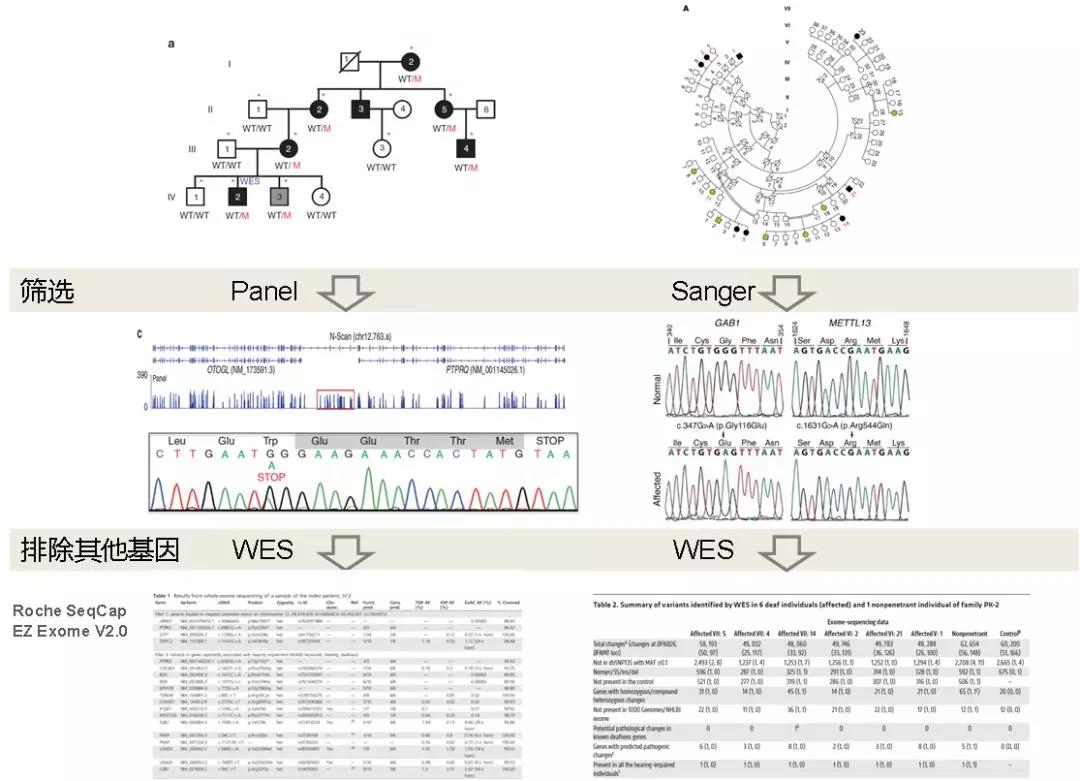
WES被認為是遺傳病致病基因發(fā)現(xiàn)的重要武器之一,Sanger測序則是作為確認WES結(jié)果的金標準,。而現(xiàn)在,,這種情況在悄然轉(zhuǎn)變。有研究表明,,對某些基因的測序結(jié)果中,,高質(zhì)量的靶向測序結(jié)果與金標準Sanger的準確性已經(jīng)旗鼓相當(dāng)[8]。在一些研究中,利用Panel或Sanger測序找到了候選致病基因后,,WES(Roche SeqCap EZ Exome V2.0)被用于排除其他基因突變致病的可能性(圖5),。時至今日,外顯子測序不再單單作為鑒定新致病基因的篩選方法,,而是逐步向多區(qū)域,、多基因高通量檢測的“金標準”邁進,腫瘤多基因Panel的接連獲批也正印證了這一點,。
去年,,繼PD-1/PD-L1抗體、CAR-T細胞治療之后,,“個性化腫瘤疫苗”在臨床治療上也取得的重大突破,。通過對腫瘤細胞進行WES和RNA-seq,篩選腫瘤細胞特異表達的突變基因,,可以為“個性化腫瘤疫苗”的腫瘤新抗原預(yù)測提供藍圖,,所以,準確的鑒定腫瘤基因突變是“個性化腫瘤疫苗”成功的基礎(chǔ),。
結(jié) 語
靶向測序的核心優(yōu)勢在于性價比高,,但不論是低頻突變檢測還是病原體鑒定,靶向測序所展現(xiàn)出的高靈敏性,,更多的是成本優(yōu)勢的間接反映,,這些任務(wù)WGS都可以完成,只不過成本過高,。值得注意的是,,靶向測序的性價比仍有提升空間。
一方面,,在完成概念驗證或數(shù)據(jù)積累后,,可以進一步精簡靶向測序的目標區(qū)間,壓縮測序成本,;例如,,在檢測TMB方面,腫瘤多基因Panel已經(jīng)被證明與WES具有較高的一致性,。在遺傳病基因診斷方面,,耳聾、癲癇等多基因Panel也有很高的診斷率,。當(dāng)然,,Panel和WES都有自己的優(yōu)缺點,也有各自的應(yīng)用場景,,Panel更加靈活,,可以根據(jù)不斷更新的數(shù)據(jù)庫,及時添加相關(guān)位點,例如一些內(nèi)含子區(qū)域,;WES多為商業(yè)化試劑盒,,往往很難做快速更新?lián)Q代,所以將“與時俱進”的Panel和商業(yè)化WES一起比較并不十分客觀,。而在應(yīng)用于表型復(fù)雜的疑似遺傳病診斷,,發(fā)現(xiàn)新基因以及重分析時,WES更具優(yōu)勢,。另一個方面,,進一步豐富靶向測序的突變檢測范圍,使之能夠同時檢測基因組SNV,、InDel,、CNV并覆蓋線粒體DNA,將有助于提升其成本效益,。
參考文獻
1. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. 2015 Nature Genetics
2. Whole genome capture of vector-borne pathogens from mixed DNA samples: a case study of Borrelia burgdorferi. 2015 BMC Genomics
3. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. 2013 NEJM
4. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. 2015 mBio
5. BacCapSeq: a Platform for Diagnosis and Characterization of Bacterial Infections. 2018 mBio
6. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. 2015 JAMA
7. Genomic diagnostics within a medically underserved population: efficacy and implications. 2017 GENETICS in MEDICINE
8. Beck TF, Mullikin JC. Systematic evaluation of Sanger validation of next-generation sequencing variants. 2016 Clin Chem
9. Modifier variant of METTL13 suppresses human GAB1–associated profound deafness,2018 JCI
10. A C-terminal nonsense mutation links PTPRQ with autosomal-dominant hearing loss, DFNA73. 2018 GENETICS in MEDICINE
11. CoDE-seq, an augmented whole-exome sequencing, enables the accurate detection of CNVs and mutations in Mendelian obesity and intellectual disability. 2018 MOLECULAR METABOLISM
來源:測序中國

